{kind=link}
{kind=link}
{kind=link}
二代测序与毛细管电泳技术STR分型差异现象探究
[郭江玲1, 2  , 尚蕾
, 尚蕾2 , 李万水2 , 丁光树2 , 杨帆2 , 孙敬2 , 孙辉2, * , 张更谦1, * ]
, 尚蕾, 张更谦]
|
|


第一作者简介:郭江玲,女,山西运城人,硕士研究生,研究方向为法医遗传学。E-mail:guojiangling1219@163.com
二代测序相较于毛细管电泳技术(capillary electrophoresis, CE),以其体系中可容纳更多的基因座而在法医学实践中更具潜力和价值。MiSeq FGxTM系统是专为法医学设计研发的一个测序平台,其配套试剂盒Forenseq DNA Signature Prep kit具有较高的灵敏度和准确度。本实验利用该试剂盒对41份家系样本进行测序,结果发现,58个STR基因座中有26个基因座出现了等位基因亚型,等位基因总数目增加了79个;与CE一致性上,有1份样本在DYS392基因座出现缺失,推测应与试剂盒引物扩增效率有关,另外还有8份样本在DXS7132基因座出现与CE不一致的情况,通过Sanger测序以及使用其他软件分析后,排除扩增失败,确认是生物信息分析问题。实验证明,二代测序相比于毛细管电泳技术有很多优势,不过目前在数据分析方面还存在一些问题,需要不断优化完善所涉及的生物信息分析方法。随着未来相关技术与标准的不断完善,二代测序会逐渐应用于法医学实践中。
, SHANG Lei, ZHANG Gengqian
Compared with capillary electrophoresis (CE) technology, next generation sequencing (NGS) can accommodate more STR genetic loci within the relevant system, therefore emerging as a promising and potential technology for forensic practice. Illumina’s MiSeq FGxTM system is a sequencing platform dedicated to forensic services, with its supporting kit - Forenseq DNA Signature Prep kit having achieved higher sensitivity and accuracy. 41 pedigree samples were here reported of being sequenced with the kit mentioned above. The results showed that 26 of the tested 58 STR loci had come into being allelic subtypes, bringing forth the total number of alleles to increase by 79. There was one sample showing a dropout at the DYS392 locus against to its CE presentation, being speculated of correlativity with amplification efficiency of the kit’s primers. Besides, eight samples were found of inconsistency with CE results at the DXS7132 locus. Sanger sequencing was thus conducted and data were analyzed with other software to explore the causation. It was confirmed that the inconsistency resulted from irrelevant bioinformatics analysis. This study has proved that NGS owns many advantages over CE although it still awaits solutions about some issues in bioinformatics analysis. With the ever-coming continuous improvement of related technologies and standards, NGS is believed to be gradually applied into forensic practice.
二代测序技术(second generation sequencing, SGS)也叫下一代测序(next generation sequencing, NGS)或大规模平行测序(massively parallel sequen-cing, MPS), 其特点是通量较大, 各个测序反应平行进行, 可以实现规模化测序, 能为法医学个案研究提供新的可能, 故正愈益成为法医遗传学中大有前途的一种方法。
相较于毛细管电泳技术, 二代测序的优势在于:
1) 体系可以容纳更多种类、更多数量的基因座。
2) 可得到详细的序列信息, 从而显著增加基因座的多态性。
3)扩增片段更短, 更适用于法医微量或降解检材[1]。
目前常用的测序平台有MiSeq FGxTM系统(Illumina, 美国)和Ion PGM测序平台(Thermo Fisher, 美国)。MiSeq FGxTM系统是专门为法医服务的一个测序平台, 其配套试剂盒Forenseq DNA Signature Prep kit包含27个常染色体STR基因座、24个Y-STR基因座、7个X-STR 基因座、94个身源识别SNP位点、22个表型SNP位点和56个地域祖先来源SNP位点。该试剂盒具有较高的灵敏度、准确度和可重复性[2, 3, 4]。
本实验分别使用CE-STR试剂盒和Forenseq DNA Signature Prep kit试剂盒对41份家系样本进行检测分析, 旨在评估等位基因序列信息对等位基因数目的增加情况并比较二代测序与传统毛细管电泳分型之间的一致性。
六个家系(图1)共41份血卡样本, 每个血卡各剪取1 cm2试样, 使用96道微量DNA提取工作站(博坤生物, 吉林长春)进行DNA提取, 得到约30 μ L的DNA溶液。用Qubit3.0(Thermo Fisher, 美国)定量至浓度在0.2~2 ng/μ L之间。
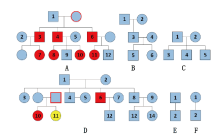
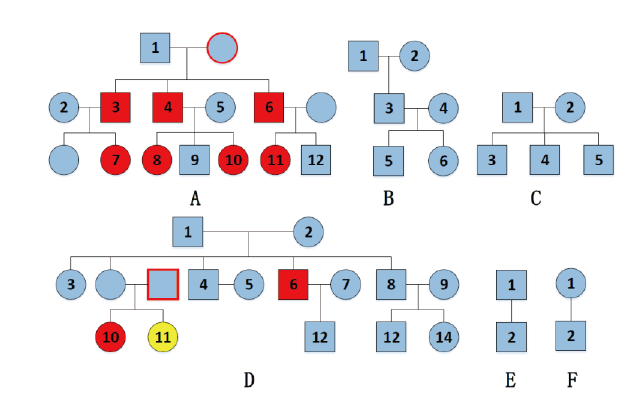 | 图1 六家系图 (各家系图下方字母为家系编号, 框内数字为样本编号, 标蓝为正常样本, 标红的为与CE结果不一致样本, 标黄的为假一致性样本, 红色外框为可能突变来源样本)Fig.1 The six pedigrees (the letters of A, B, C, D, E, F representing each pedigree selected, with the enclosed number indicating each sample as blue: normal samples; red: samples whose NGS results are inconsistent with the CE ones; yellow: false consistent samples; framed red: possible source sample having caused mutation) |
本研究经公安部物证鉴定中心科研伦理委员会审查通过, 符合要求。样本提供者均签署了知情同意书。
使用DNATyperTM21、DNATyperTMY36、DNATyperTMX19试剂盒(均为公安部物证鉴定中心产品)对所有样本按说明书进行扩增检测, 扩增产物以3730XL型基因分析仪(Thermo Fisher, 美国)电泳检测, 采用GeneMapper ID-X 1.5进行分析。对NGS与CE结果不一致的样本加做Investigator Argus X-12 QS试剂盒(QIAGEN, 德国)验证。
将所有样本DNA稀释至0.2 ng/μ L, 按照Forenseq DNA Signature Prep kit试剂盒说明书构建文库, DNA模板输入量为1 ng。扩增遗传标记包括27个常染色体STR、24个Y-STR、7个X-STR和94个iSNP。使用MiSeq FGxTM Reagent Kit试剂盒Micro Flow Cell芯片, 按说明书在MiSeq FGxTM测序仪上进行测序, 原始数据以Forenseq UAS(Illumina, 美国)在默认分析阈值下处理。对其中不一致样本使用方明生物信息分析平台(北京中科方明科技公司)进行二次分析, 分析阈值和解释阈值分别为2%、5%。
对家系D中所有男性样本的DYS392基因座进行Sanger测序(生工生物, 上海); 对DXS7132基因座二代测序与CE分型结果不一致的8份样本以及分型一致的3份样本(A-2、D-5、D-11)也进行Sanger测序。
两次测序的簇密度(cluster density)分别为1 553K/mm2、1 335 K/mm2(推荐范围为400~1 650 K/mm2); 簇通过率(cluster passing filter)分别为86.08%、89.88%(推荐范围为≥ 80%); 定向值(phasing)分别为0.144%、0.171%(推荐范围为≤ 0.25%); 预定向值(pre-phasing)分别为0.162%、0.063%(推荐范围为≤ 0.15%)。其中第一次测序预定向值超出推荐范围, 或因簇密度较高导致, 其余参数均在推荐范围内, 总体测序质量较好。
二代测序能够获得序列信息, 可以发现更多的等位基因“ 亚型” [5]。本次实验检测41个样本, 所测58个基因座共有2 378个等位基因的序列分型, 其中有26个基因座发现了等位基因亚型, 等位基因数目增加情况如表1, 以D12S391增加最为显著, 由9个增加到18个, 对于常染色体STR以及X-STR、Y-STR基因座, 等位基因总数分别由204、45、91增加到了265、51、103, 共增加了79个。
| 表1 41个样本中STR基因座等位基因数目增加情况 Table 1 Occurrence to increased number of STR alleles among 41 samples |
2.3.1 分型不一致样本情况及分型
58个STR基因座中有4个基因座(D2S441、D10S1248、D22S1045、DXS10074)无CE结果, 对其余基因座进行一致性比较, 发现41个样本中有9个样本出现与CE结果不一致的情况, 如表2所示。
| 表2 二代测序与CE结果不一致情况比较 Table 2 Comparison of inconsistent STR genotypes between NGS results and CE ones |
2.3.2 DYS392基因座分型差异分析
D-6在DYS392出现等位基因丢失, 使用方明分析平台重新分析同样出现丢失, 而测序结果未发现序列差异, 相关文献对该基因座均有类似报道[6, 7, 8, 9], 等位基因越大时, 等位基因数会越低甚至低于阈值。因此, 考虑可能为试剂盒引物扩增效率问题。使用自行研发的高通量测序体系检测时未发生等位基因丢失现象。
2.3.3 DXS7132基因座分型差异分析
基因座DXS7132在样本A-3、A-4、A-6、A-7、A-8、A-10、A-11、D-10中显示与CE结果不一致, 出现了等位基因丢失的情况。通过Investigator Argus X-12 QS试剂盒验证, 结果与DNATyperTM X19试剂盒一致, 如图2a、2b。A家系中有多个样本出现等位基因12的丢失, 结合家系图(图1)进行分析, 考虑可能存在家族遗传倾向, 推测A家系中祖母存在突变; 而D家系中同胞姐妹D-10、D-11的父母样本缺失, 推测父亲可能存在突变, 同时D-11可能为假纯合子, 如表3所示。
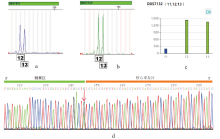
 | 图2 四种方法对样本A-8 DXS7132基因座的分型结果 (a:DNA TyperTM X19; b:Investigator Argus X-12 QS; c:方明分析平台; d:Sanger测序)Fig.2 Genotyping results of DXS7132 from sample A-8 with four methods (a: DNA TyperTM X19; b: Investigator Argus X-12 QS; c: ForemicsTM software; d: Sanger sequencing) |
| 表3 D-10、D-11突变来源分析 Table 3 Analysis of mutation origin into D-10 and D-11 |
选取所有不一致样本以及两份一致样本进行Sanger测序, 通过与参考序列比对发现仅在重复区域下游第一个碱基发生G/A突变, 如图2d, 其余位置均与参考序列一致, 该突变不在引物结合区, 因此排除扩增失败的原因。随后将原始测序FastQ数据用方明生物信息分析平台进行分析, 得到了与CE相一致的结果, 如图2c。因此, DXS7132基因座等位基因的缺失并非PCR扩增失败, 而是生物信息分析问题。而对于样本D-11, 通过查找二代测序侧翼序列报告, 证明该样本Forenseq UAS分析结果缺失了来自父方的等位基因13, D-11的正确分型如表4所示。
| 表4 样本D-11的正确分型 Table 4 The correct genotyping of sample D-11 |
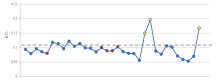
将实验中所有女性样本DXS7132的每条等位基因reads数占X-STR总reads数的比值绘制成折线图, 如图3, 其中蓝色点为杂合子样本每条等位基因reads数的占比, 红色、黄色和绿色点为纯合子样本的reads数占比, 发现红色、绿色点样本的占比与蓝色基本一致, 提示其可能是杂合子的其中一条等位基因, 意味着其中一条等位基因的丢失。红色点正是出现等位基因丢失的样本, 而样本D-11(绿色点)虽然与CE的结果一致, 但通过折线图也可以发现缺少了一条等位基因, 通过这种方法可以发现NGS中假纯合子的情况, 故诚可增加NGS分析的准确性。
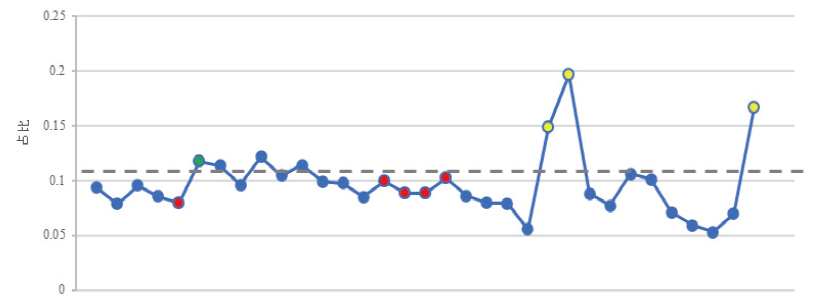 | 图3 所有女性样本DXS7132基因座等位基因reads数与X-STR总reads数比值 (图中蓝色点表示杂合子样本每条等位基因的reads数占比, 红色、黄色和绿色表示纯合子样本reads数占比, 其中红色表示与CE结果不一致, 绿色表示与CE结果假一致, 黄色表示与CE结果一致)Fig.3 Ratio of allelic reads at DXS7132 locus to total X-STR reads from all female samples (The blue dots representing the percentage of reads per allele on its residing chromosome in heterozygous samples, with the red, yellow and green dots describing the percentages of allelic reads in homozygous samples as Red: inconsistent with CE’ s; Green: falsely consistent with CE’ s; Yellow: consistent with CE) |
二代测序结果生物信息分析的一种策略, 是通过特异序列比对查找相应基因座的序列, 当这些位置发生突变时, 可能导致这些基因座等位基因的丢失, 本次实验中DXS7132基因座不一致的原因仅仅是3’ 侧翼一个碱基的突变。Wang等[10]的实验观察到1份样本在D7S820基因座、2份样本在D21S11基因座出现等位基因的丢失, Sanger测序结果发现在侧翼区出现碱基插入, 通过NextGENe® 软件重新分析后, 得到了正确的分型。Barrio等[11]在使用Converge 2.0软件进行分析时, 有3份样本在Penta D基因座出现等位基因2.2的丢失, 当使用STRait Razor和Integrative Genomics Viewer (IGV) v 2.4.16重新分析后, 得到了与CE相一致的结果, 分析发现是5’ 侧翼区出现13 bp的缺失。这些与CE分型结果不一致的情况, 往往发生在数据分析阶段, 通过更换其他不同的分析软件(如STRait Razor、NextGENe® 等)可以得到纠正。
除了生物信息分析的原因, 二代测序与CE分型差异的原因还包括引物结合区突变、核心区长度计算方法差异、测序错误等。首先, 引物结合区发生突变会造成等位基因扩增失败, 从而导致CE与二代测序结果的差异。Kwon等[12]的实验中, 1份样本在DYS439基因座NGS结果为12/13, 使用Powerplex Y23和荧光标记的NGS引物分别进行毛细管电泳, 得到的分型结果分别为13和12/13, 不一致的原因是引物结合区域的突变。Xue等[13]在D8S1179基因座观察到了等位基因16的丢失, 原因是反向引物结合区出现G/A突变。其次, 毛细管电泳技术只能检测扩增产物的长度, 传统的核心重复区长度的计算方法是产物长度减去固定的侧翼长度, 但是当侧翼区域出现插入或缺失时, 就会出现CE与NGS结果不一致的情况。Barrio等[11]通过二代测序在对496份无关个体的31个常染色体STR进行检测时发现, 其中1份样本的D19S433基因座CE的分型为13.2/14, 而NGS结果为纯合子14, 其原因是侧翼区发生2 bp碱基的缺失。因此, 二代测序实际上可以得到更为准确的结果, 但同时也会导致与毛细管电泳不一致的结果。最后, 测序过程中的测序错误也会导致错误分型, 从而出现与CE不一致的结果。Liu等[14]的实验中, FGA基因座的CE结果为24/26, 二代测序结果中, Converge软件分析结果为19.3/26, STRait Razor软件分析结果为24, 通过克隆测序发现, 核心重复区下游的一段序列“ TTTCTTTTTT” , 用二代测序会出现错误, 产生“ TTTCTTTTTT” 和“ TTTTCTTTTT” 两种测序结果, 等位基因24、26产生的401和218个reads中, 有218和38个reads测序正确, 因而在生物信息分析时, 就出现错误的结果。
实际上, 引物结合区突变、核心区长度计算方法差异以及样本侧翼区序列变化导致分型结果不一致的情况也出现在CE-STR试剂盒之间。Hill等[15]在比较试剂盒AmpFlSTR MiniFilerTM与Identifiler STR kits的一致性时发现27例不一致情况。Huaxia Platinum CEF2 Kit、SinofilerTM等试剂盒也有类似报道[16, 17, 18]。这种分型差异在实际工作中同样要引起注意。
2020年, 刑事技术标准化技术委员会发布了《法庭科学DNA二代测序检验规范》(GA/T1693-2020) 和《序列多态STR等位基因命名规则》(GA/T1694- 2020), 随着未来命名标准和数据分析方法的进一步完善统一, 二代测序与毛细管电泳之间分型差异的现象会得到改善。概言之, 二代测序相较于毛细管电泳技术有很多优势, 其在法医学鉴定中的应用会越来越广泛。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|