
{kind=link}
{kind=link}
运用二代测序技术拆分混合STR分型
[吴玉剑1  , 方慧
, 方慧1 , 刘翠兰1 , 韦甜2 ]
, 方慧|
|


第一作者简介:吴玉剑,男,四川广元人,博士,工程师,研究方向为法医遗传学。E-mail: scmirror@163.com
目的 探讨二代测序技术在混合STR分型拆分中的应用。方法 对一例强制猥亵妇女案中受害人颈部的拭子和血样作DNA提取,以Precision ID GlobalFilerTM NGS STR Panel v2试剂盒制备文库,经Ion S5测序仪测序,运用Torrent_Suite_v5.2.1软件进行数据分析后,将检测基因座的序列多态STR分型与长度多态STR分型进行比较。结果 在D8S1179、D21S11、D2S441、D2S1338、D10S1248五个基因座发现存在序列特异的等位基因亚型,利用这些亚型对混合STR分型进行了成功拆分。结论 二代测序技术提供的等位基因序列信息可对混合STR分型的拆分起到帮助作用。
Objective To explore the applicability of next generation sequencing (NGS) into STR (short tandem repeats) genotyping of mixed DNA.Methods DNA was extracted from one sex-harassed woman with both the swab of wiping the woman´s neck and blood samples that were collected through the case. The Precision ID GlobalFilerTM NGS STR Panel V2 was used to prepare the library that was subsequently sequenced by the Ion S5 sequencer. The data were analyzed by Torrent_Suite_v5.2.1 software, with the relevant STR profiles being compared between the sequence- and length-based STR ones.Results Sequence-specific allelic subtypes were found in five loci: D8S1179, D21S11, D2S441, D2S1338 and D10S1248, thereby having made the relevant mixed STR profiles successfully separated.Conclusion The allelic sequence information parsed through NGS can assist in separating mixed STR profiles.
随着DNA检测灵敏度提高, 从体液、分泌液以及脱落细胞类型的检材中检出混合STR分型的几率不断增加, 如何从混合STR分型中发掘出有价值的信息也成了某些案件侦破的关键[1, 2, 3]。传统的混合STR分型拆分的方法主要参考等位基因个数、峰面积等信息[4], 依赖于技术人员的经验。然而由于影子带、拔起峰、非特异性带、扩增不平衡等因素的影响[5, 6], 混合STR分型的拆分一直难有很好的解决方案, 如何科学准确地对混合STR分型进行拆分就成了法医物证领域的一个难点和研究方向。本文通过Ion S5二代测序平台对一例二人混合DNA样品进行测序分析, 并利用等位基因序列信息对部分基因座混合STR分型进行了成功拆分。
2018年12月, 我市某小区发生一起强制猥亵妇女案, 技术人员提取了受害人颈部擦拭拭子、受害人血样。
PrepFiler Express BTATM试剂盒、GlobalFilerTM试剂盒、Precision ID GlobalFilerTM NGS STR Panel v2 引物试剂盒、Ion AmpliSeq Kit for Chef DL8文库构建试剂盒、Ion 520 & Ion 530 Kit-Chef模板制备试剂盒、Ion S5 Sequencing Kit测序试剂盒、Ion 520™ Chip型芯片、AutoMate ExpressTM纯化仪、9700型扩增仪、3500 xL基因分析仪、Ion S5测序仪、7500 型实时定量扩增仪、Ion Chef工作站, 来自美国Thermo Fisher公司。
使用AutoMate ExpressTM纯化仪(AB公司)及PrepFiler Express BTATM试剂盒进行DNA提取, 使用 GlobalFilerTM 试剂盒进行STR 复合扩增, 扩增条件为:95 ℃、1 min; 94 ℃、10 s, 59 ℃、90 s, 29个循环; 60 ℃、10 min, 4℃保存。使用3500 xL基因分析仪和GeneMapper® ID-X软件进行电泳和数据分析。
使用AutoMate ExpressTM纯化仪(AB公司)及PrepFiler Express BTATM试剂盒进行DNA提取, 使用Precision ID GlobalFilerTM NGS STR Panel v2和Ion AmpliSeq Kit for Chef DL8试剂盒构建文库, 使用Ion 520 & Ion 530 Kit-Chef试剂盒制备模板, 使用Ion S5 Sequencing Kit和Ion 520™ Chip型芯片以Ion S5测序仪测序; 使用Torrent_Suite_v5.2.1软件对测序原始数据进行初步筛选及分析, 使用Coverage_Analysis和 HID_STR_Genotyper插件以Hg19基因组为参照对测序结果进行综合分析和STR分型检测。
使用GlobalFilerTM试剂盒结合毛细管电泳平台从受害人颈部拭子中提取的DNA检测到二人混合STR分型, 包含受害人DNA分型和未知男性DNA分型(图1和表1)。为了对该混合分型结果做进一步验证分析, 继续使用二代测序的方法对颈部拭子DNA进行检测。本次使用的Precision ID GlobalFi-lerTM NGS STR Panel v2 试剂盒中包含31个常染色体STR基因座和4个性别相关基因座, 覆盖了GlobalFilerTM试剂盒中除SE33外的全部基因座。结果显示二代测序检测的基于长度多态性STR分型与毛细管电泳STR分型完全一致(表1), 两种方法检测的结果可以互相印证。
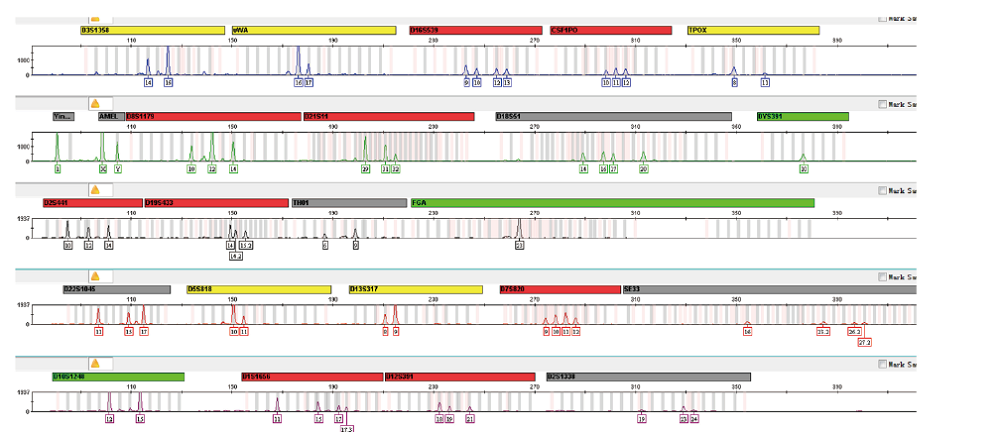 | 图1 颈部拭子常染色体STR基因座分型图谱Fig.1 Autosomal STR profile from the swab wiped of the victim’ s neck where the kissing was coercively conducted |
| 表1 颈部拭子二代测序STR分型和受害人STR分型 Table 1 NGS-revealed STR loci from the swab of wiping the victim’ s neck and CE-exposed loci from the victim’ s blood |
2.2.1 利用核心序列差异辅助拆分混合STR分型
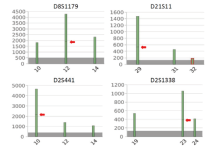
二代测序数据显示在D8S1179基因座的等位基因12、D21S11基因座的等位基因29、D2S441基因座的等位基因10和D2S1338基因座的等位基因23上均存在两个等位基因亚型, 其核心序列长度相同但是碱基序列不同(图2)。借助等位基因序列多态性可以对上述基因座的混合分型进行拆分。在D8S1179基因座上, 长度多态性STR分型为10/12/14, 受害人分型为10/14, 未知男性分型可能是12、10/12或者12/14, 借助序列多态性可知该位点等位基因12由两个不同的亚型组成, 由于受害人无等位基因12, 所以这两个亚型必然由未知者提供, 因此可以判断此未知者在该处分型为12。在D21S11基因座上, 长度多态性STR分型为29/31/32, 受害人分型为29/31, 未知者分型可能是32、29/32或31/32, 借助序列多态性可知该位点等位基因29由两个不同的亚型组成, 受害人只能提供一个亚型, 另一个亚型必然来自未知者, 因此可以判断未知者在该处的分型为29/32。基因座D2S441、D2S1338和D21S11情况类似, 同理可以拆分出未知者在这两个基因座的分型分别为10/12和23/24。
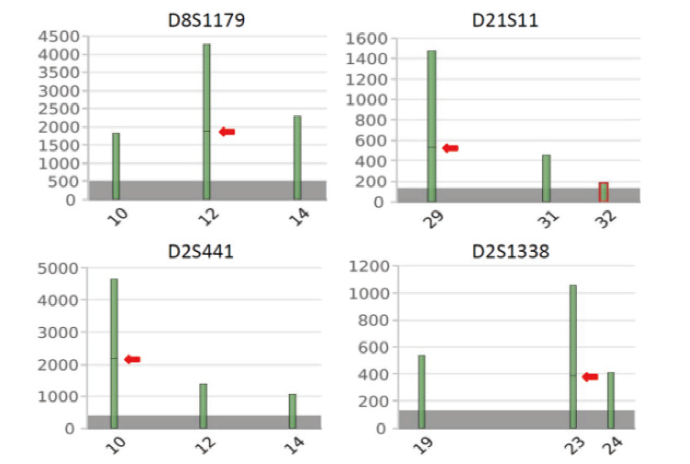 | 图2 颈部拭子DNA在D8S1179、D21S11、D2S441、D2S1338基因座的NGS-STR分型Fig.2 NGS-detected STR genotypes of D8S1179, D21S11, D2S441 and D2S1338 loci from the swab of wiping the victim’ s neck |
2.2.2 利用侧翼序列差异辅助拆分混合STR分型
在基因座D10S1248上, 二代测序结果显示无论是基于核心序列长度多态性还是基于序列多态性, 其STR分型都是12/15, 与毛细管电泳STR分型一致。但是通过分析等位基因12核心序列两边的侧翼序列发现等位基因12是由两个不同的亚型组成, 12a和12b(表2)。等位基因12a在侧翼序列chr10:131092586处为C碱基, 而等位基因12b在该位置的C碱基缺失。受害人在该基因座上的分型为12/15, 只能提供等位基因12的一个亚型, 另一个亚型必然来自未知者, 即未知者在该基因座的分型一定包含等位基因12。
| 表2 混合DNA在D10S1248基因座上等位基因12的测序信息 Table 2 Genetic information of allelic 12a and 12b at D10S1248 locus from sequencing into the mixed DNA sample |
传统的毛细管电泳STR分型技术只能检测STR基因座的长度多态性信息, 不能充分挖掘DNA中蕴藏的遗传信息。二代测序既能检测STR基因座的长度多态性信息, 又能分析各基因座序列多态性信息, 通过分析核心序列和侧翼序列的碱基序列差异, 可对等位基因进行精准分型, 故在法医物证领域发挥着越来越重要的作用[7, 8, 9, 10, 11]。本文使用Precision ID GlobalFilerTM NGS STR Panel v2试剂盒在Ion S5平台上对一例混合DNA样品进行了测序分析, 得到的长度多态性STR分型与CE-STR分型一致。另外, 在D8S1179、D21S11、D2S441和D2S1338 基因座观察到核心序列变异, 发现其长度相同而碱基序列不同。这些核心序列变异可以分为2类[12, 13]:一类是核心序列重复单元出现数目变异, 例如D2S1338基因座等位基因23分为[TGCC]8[TTCC]12 GTCC[TTCC]2和[TGCC]7 [TTCC]13 GTCC[TTCC]2; 另一类是核心序列重复单元出现SNP, 例如D2S441基因座的等位基因10分为[TCTA]10和[TCTA]8 TCTG[TCTA]1。在测序过程中等位基因核心序列已经被测序完全覆盖, 无法通过调整测序策略获得更多的核心序列信息, 此时检测侧翼序列遗传标记可作为重要的补充手段。将更多的单核苷酸多态性(SNP)、插入/缺失(InDel)位点纳入侧翼序列检测范围, 与STR位点组合成联合遗传标记, 可能是提高混合分型拆分能力的一个有效途径。使用CE-STR技术对混合分型拆分的方法受混合组分的比例影响很大, 本研究中受害人和未知男性DNA含量接近, 借助受害人分型按照传统方法对所研究的几个基因座混合分型进行拆分并不困难, 但是当混合比例差异比较大, 未知男性DNA混合比例很低时, 由于影子带、拔起峰等因素的影响, 传统方法可能就无法对这几个基因座进行准确的识别和拆分。二代测序直接读取等位基因序列, 可以排除干扰, 受混合组分比例的影响小, 有研究表明混合比例低至1:19时仍能检测到次要组分的部分等位基因[12]。二代测序不但可以对等位基因亚型进行精细分型, 同时还可以根据测序数据量对等位基因进行定量分析, 从而较为准确地确定混合组分之间的比例, 对混合分型拆分起到帮助作用。
本文使用二代测序技术检测了混合样品的31个常染色体基因座, 只在文中描述的5个基因座检测到存在等位基因亚型, 所占比例较低, 也显示出目前该方法在混合分型拆分方面的不足之处。然而, 在实际案件中, 一些特殊基因座(如包含4个等位基因)借助受害人的分型按照传统方法可以准确拆分出未知者分型, 这时再利用二代测序技术额外拆分出几个基因座分型, 就可以得到足够的位点信息进行同一认定或者进行数据库检索, 起到重要的补充作用。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|